1 University Clinic for children’s diseases, department of Neurology, Skopje, North Macedonia.
2 University Clinic of Neurology, Skopje, Macedonia.
*Corresponding Author:
Alili Ademi L, University Clinic for children’s diseases, department of Neurology, Skopje, North Macedonia.
Citation:
Alili Ademi L, Duma F, and Ademi B (2025), Electro-Clinical Pattern of Epilepsy in A Child with ANKRD11 Mutation
Associated with KBG Syndrome: Case Report of Drug-Resistant Epilepsy; J. Neurology and Neurological Research, 2(1): DOI: SH-NNR-CR-006.
Introduction: Epilepsy is the most common neurological disorder in the pediatric population, of different etiology. 30% of children have drug-resistant epilepsy affecting their cognitive function, leading to worsening of the prognosis, serious psychosocial consequences, difficulties in care and quality of life, anxiety in the family, as well as an increase in the risk of death. Idiopathic ones, have a genetic basis. ANKRD11 genetic mutation is associated with epilepsy. ANKRD11 is the main gene responsible for KBG syndrome, characterized by seizures, delayed closing of fontanels, and electroencephalographic (EEG) abnormalities. Up to date a systemic review of epilepsy and EEG abnormalities with KBG syndrome is not sufficient.
Aim: This case report aims to contribute to identifying a specific electroclinical pattern of KBG syndrome.
Case report: We report a patient presenting with a drug-resistant, monogenic epilepsy syndrome, due to ANKRD11 pathogenic de novo variant found by TES associated with the KGB syndrome. We report the electroclinical pattern of epilepsy associated with KBG and outline the clinical features of the epileptic seizures and EEG features of this patient.
Conclusion: The boy suffered from focal to tonic-clonic seizures, treated with various AEDs for seizure control. Even though our result cannot be conclusive, we hypothesize that the represented EEG pattern may help to characterize the phenotype of KBG syndrome.
Key Clinical Message: EEG pattern together with the epilepsy and seizure type, dysmorphic facial features, ID, and behavioral disorders, may help to characterize the phenotype of KBG syndrome.
INTRODUCTION
Epilepsy is a common neurological disorder affecting more than 70 million people around the world. It is the most common neurological disorder in the pediatric population, affecting 1% to 3% of children, and is defined as any disorder in which spontaneous recurrence of unprovoked seizures is the main symptom. A seizure is usually defined as a sudden alteration of behavior due to a temporary change in the electrical functioning of the brain, that continuously generates tiny electrical impulses in an orderly pattern. The etiology of epilepsy can be different, including structural, genetic, infectious, metabolic, and immune causes, but most often the cause is unknown, which occurs in 40% of people with epilepsy. Despite the recent introduction of new antiepileptic drugs (AEDs), about one-third of epilepsy patients have drug-resistant (refractory) epilepsy, affecting about 30% of children with epilepsy.
Refractory epilepsy, which is the most severe form of epilepsy, according to the International league against epilepsy (ILAE), is defined as failure to control seizures when using two or more appropriately chosen and tolerated antiepileptic drugs (as monotherapy or in combination) during an appropriate period. Severe and refractory epilepsies in children affect their cognitive function, leading to worsening of the prognosis, serious psychosocial consequences, difficulties in care and quality of life, anxiety in the family, as well as an increase in the risk of death, including unexpected death in epilepsy (SUDEP).
The expansion of genetic research and technologies in recent years and the advances in next-generation sequencing (NGS) have shown that a large proportion of unexplained epilepsies, especially idiopathic ones, have a genetic basis. Numerous epilepsy genes helped the understanding of mechanisms underlying epileptogenesis and guided the development of treatments.
ANKRD11 gene encodes the protein ankyrin repeat domain 11 (ANKRD11), which isfound in the neurons in the brain. The ANKRD11 protein functions as a co-regulator in the developing brain, playing a critical role for the neural proliferation, for the genesis and positioning of newborn neurons, for neuronal plasticity (ability of neurons to change and adapt over time), which is important for learning and memory.
The role of the ANKRD11 gene in neurodevelopment was suggested by subsequent reports of individuals with intellectual disability, facial dysmorphism, and ASD. In 1975, Herrman et al., first described the KBG syndrome, a neurodevelopmental autosomal dominant disorder, caused by the haploinsufficiency of the ANKRD11 at 16q24.3 locus due to heterozygous pathogenic variants or chromosomal imbalances/rearrangement such as point mutations, duplications or microdeletions involving this gene.
KBGS is characterized by global developmental delay (DD), intellectual disability (ID), learning difficulties, neurobehavioral problems, short stature, macrodontia, facial dysmorphism, skeletal anomalies, and multiple congenital anomalies, sometimes associated with seizures, delayed closing of fontanels and electroencephalographic (EEG) abnormalities. So far, there have been reported more than 200 cases of KBG syndrome.
The diagnosis of KBG syndrome is established, by the most commonly used criteria of Skjei et al. and Low et al. A diagnosis of epilepsy, a major criterion, according to Skjei et al., and a minor criterion according to Low et al. has been reported in approximately 30% of patients in a systematic review of 140 patients with KBG syndrome. The onset of epilepsy is predominantly between infancy and mid-teens and seizure remission occurred in the majority after adolescence with good response to AEDs. Heterogeneous seizure types have been reported, with tonic–clonic seizures, absences, myoclonic seizures, and unclassified sleep-related seizures with motor symptoms being most common, but no specific epilepsy syndrome has been identified.
Detailed classification of the seizures, epilepsy type, or syndrome was only made in 26 patients from 12 studies. Only two patients had an epilepsy syndrome classification. No genotype–phenotype correlation studies have been performed for the presence and type of epilepsy in patients with KBG syndrome. Generalized epilepsy with febrile seizures plus (GEFS+) is reported in a de novo mutation of the ANKRD11 gene, with a clinical phenotype compatible with KBG syndrome.
A systemic review of epilepsy and EEG anomalies in subjects with KBG syndrome is deficient. Samanta first described in a patient an intermittent bisynchronous temporo-occipital rhythmic delta activity and episodes of staring spells with no EEG changes suggesting that these findings may be specific to KBG syndrome. Here, we report a patient with a severe neurological phenotype of KBG syndrome associated with a novel heterozygous frame-shift de novo variant in the ANKRD11 gene, to contribute to identifying a specific electroclinical pattern of KBG syndrome.
CASE REPORT
We report on a 16-year-old boy, referred to our department for afebrile unprovoked seizures. He was born at term by spontaneous vaginal delivery, of healthy unrelated parents, and with uneventful perinatal and postnatal clinical course. He did not experience febrile convulsions, traumatic brain injury, or central nervous system infections. Over time he was found to have a mild global learning difficulty that improved over time with occupational therapy. He was reported by the parents to be a very shy, quite timid, reticent, and solitary person. Also, stereotype and behavioral changes were noted. Emotional and social difficulties were reported by the family. During clinical assessment, craniofacial dysmorphic features of KBG syndrome were noted. Regarding the neurologic and behavioral aspects, the patient had mild intellectual disability, learning difficulties, and neurobehavioral problems such as attention deficit hyperactivity disorder (ADHD) since childhood. However, he was able to participate in normal school life without any help.
Targeted exome sequencing (TES) of 4800 clinically significant genes was performed and a pathogenic heterozygous variant, NM_001256183.2: c.4407dupG, frameshift, was identified in ANKRD11, c.4407dupG, (p. Arg1470GlufsTer84). As the parental genetic tests revealed that the parents did not have this frameshift, the variation was identified as a de novo variant. This variant was categorized as pathogenic by the American College of Medical Genetics and Genomics (ACMG) guideline (PVS1, PS1, PS2, and PM2) and has not been reported earlier. The variant is absent from control sequences (Genome Aggregation Database (gnomAD) and no alternative plausible variants or known mutations were identified as competing possibilities in this patient.
Seizure type
EEG abnormalities
Age
AEDs
Seizure frequency
GTCS
Multifocal discharges
8 yo
VPA
First seizure
GTCS +
FS
Focal/ multifocal bursts
9 yo
VPA+ LTG (ex, ar)
+ ACZ
1-2 /month
Stabilization
No abnormalities
9-11 yo
VPA + ACZ
GTCS
One FS
Focal bursts
11 yo
VPA+ ACZ + LEV
1-2 /week
GTCS
Focal /multifocal bursts
12 yo
VPA + ACZ + LEV
1/month
FS
Multifocal paroxysmal discharges
13-15 yo
VPA + ACZ + LEV
LEV – ex
1/week
GTCS
Multifocal bursts
15 yo
OXC (ex,ar)+ TPM
VPA – ex
ACZ – ex
1/week
GTCS
Multifocal bursts
16 yo
TPM - ex
+ LTG
2-3/month
GTCS
Multifocal bursts
nowadays
LTG
2-3/year
Table 1: Electroclinical characteristics of epilepsy
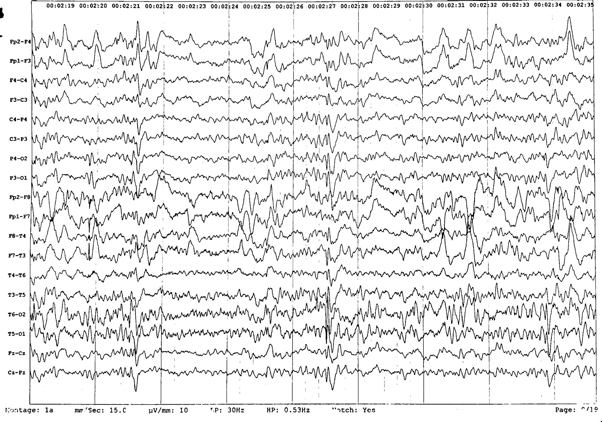
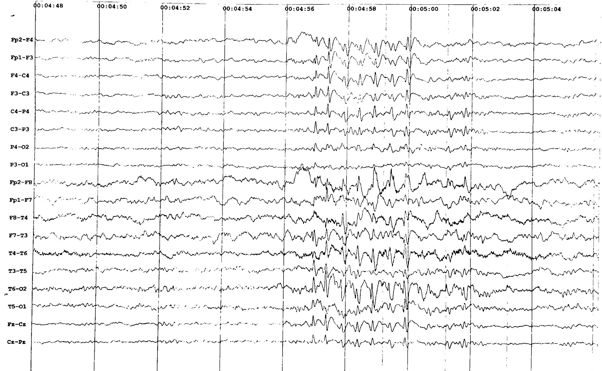
His first seizure was a generalized tonic-clonic seizure by description and occurred at the age of 8 years, when he was admitted in our clinic, department of neurology. The first wakefulness interictal EEG showed regular alpha activity, associated with occasional multifocal paroxysmal bursts of high voltage spike-wave complexes (see picture 1). Treatment with sodium valproate was started. Initially, there were 1-2 generalized tonic-clonic seizures per month on average, by the age of 9.
Figure 1. Wakefulness interictal EEG showing occasional multifocal paroxysmal bursts of high voltage spike-wave complexes.
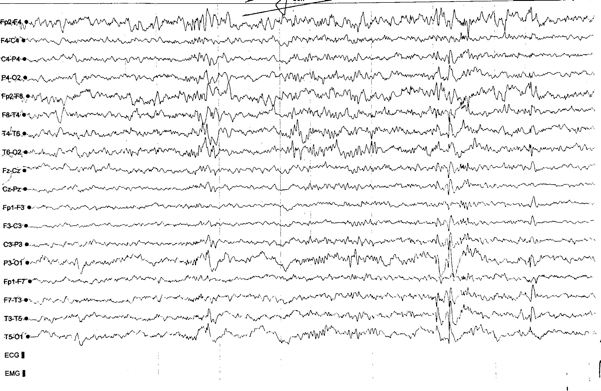
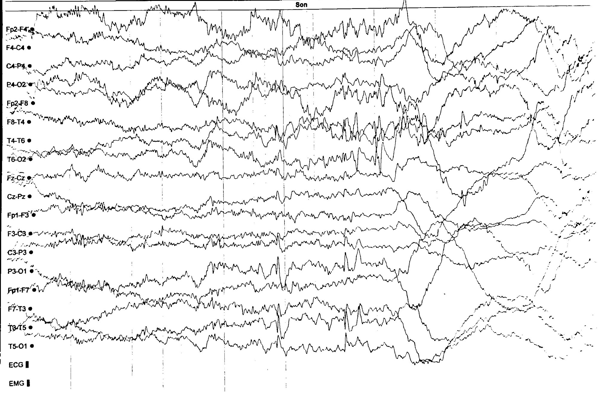
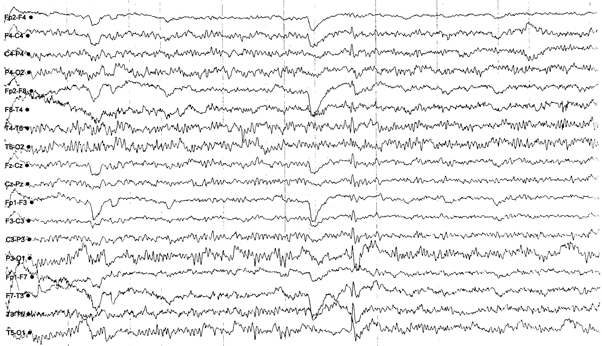
Stabilization of seizures was not achieved, there were also focal seizures. Therefore, a second AED, lamotrigine, was added to the treatment. Regarding the allergic reaction due to lamotrigine, it was discontinued, and the treatment included acetazolamide. During this period the EEG findings in wakefulness and post-hyperventilation, showed occasional multifocal mainly bitemporal bursts of spike-and-waves, while on sleep EEG revealed occasional multifocal left-sided frontotemporal and parietooccipital spike-and-waves bursts (see pictures 2, 3, 4).
A period of seizure freedom occurred from age 9 to 11 occurred after commencing acetazolamide. There was no history of absence seizures or myoclonic seizures, meanwhile, episodes of staring spells with no EEG changes were noted. The follow-up wakefulness EEG findings showed stabilization, meaning a regular brain activity of alpha rhythm with isolated right-sided spike-and-wave bursts.
Thereafter, followed a period of frequent twice-a-week generalized tonic-clonic seizures and one focal epileptic seizure. Levetiracetam was included in the antiepileptic treatment. Repeat wakefulness EEG findings showed focal bursts of left-sided parieto-occipital and temporal spike-wave discharges. (see picture 5). Epilepsy protocol magnetic resonance imaging of the brain was normal.
Figure 5. Wakefulness EEG findings showing focal bursts of left-sided parieto-occipital and temporal spike-wave discharges.
During a period of a year, the boy had one tonic-clonic generalized seizure per month, and no focal seizures were noted. The wakefulness EEG findings revealed basic brain activity of regular alpha rhythm with intermittent photic stimulation and hyperventilation activation of focal bursts of frontal and temporal spike-and-waves.
A round two-year period of focal seizures followed, and the wakefulness EEG findings showed regular alpha activity with multifocal bilateral paroxysmal discharges of spike-and-wave 2 c/s, lasting 3-4 seconds (see picture 6).
The patient was not seizure-free for a long period of time. He had tonic-clonic generalized epileptic seizures once a week in the following years. In the epileptic treatment another AED, oxcarbazepine, was added. The EEG findings during this period showed focal bursts of right-sided spike-and-wave. After a short period of time, exactly a two-week period, oxcarbazepine was excluded, due to allergic reaction regarding the drug, and topiramate was added. Valproate was excluded from the treatment.
The period that followed was characterized by 2-3 tonic-clonic generalized epileptic seizures per month. Topiramate was excluded and lamotrigine was added. No allergic reaction was noted. For a short period of a few months, he was seizure-free. Now the patient is on treatment with lamotrigine, he is not seizure-free completely, but stabilized and has only a few seizures per year. The follow-up wakefulness EEG findings showed regular beta rhythm with right-sided multifocal bursts of spike-and-slow waves.
His seizures proved difficult to control from age 12 when generalized tonic-clonic seizures recurred reaching a frequency of up to 2-3 seizures per month, despite trials of lamotrigine, levetiracetam, and topiramate. A trial of topiramate in combination with lamotrigine led to a marked reduction in seizure frequency, reducing to three generalized tonic-clonic seizures in the year following treatment initiation.
DISCUSSION
Epilepsy and EEG abnormalities are frequently reported in KBG patients. Most often present with generalized or combined generalized and focal epilepsy with onset in childhood. Literature data are limited, incomplete, and inconclusive to identify a specific electroclinical pattern. Various seizure types have been reported in the literature, with tonic-clonic generalized seizures being the most frequent seizure types. Focal seizures are also rarely noted. Moreover, many cases of generalized and focal EEG abnormalities without clinically evident seizures have been reported. In most of the KBG syndrome patients with epilepsy, seizures are drug-resistant. Moreover, epilepsy in patients with KBG syndrome is associated with poorer developmental outcomes.
We report a boy presenting with a drug-resistant, monogenic epilepsy syndrome, due to ANKRD11 pathogenic de novo variant found by TES associated with the KGB syndrome. This variant has never been reported so far in the literature. The reassessment of phenotypic features confirmed that he fulfilled the proposed diagnostic criteria for KBG syndrome.
CONCLUSION
We outline the electroclinical pattern of epilepsy in KBGs, types of the epileptic seizures and EEG features of this patient. He suffered from focal to tonic-clonic seizures, treated with various AEDs, making a drug-resistant epilepsy diagnosis. Even though our result cannot be conclusive, we hypothesize that the represented EEG pattern together with the epilepsy and seizure type, dysmorphic facial features, ID, and behavioral disorders, may help to characterize the phenotype of KBG syndrome. Future studies regarding these issues may outline the electroclinical pattern in a larger series of patients with KBG syndrome. Moreover, further research on genotype-phenotype correlations in large number of patients with KBG syndrome is encouraging.
AUTHORS CONTRIBUTIONS
LAA had main contribution in literature search, writing and drafting the manuscript. All authors contributed in the diagnosing, treatment, and follow‐up of the patient, edited the manuscript. All authors approved the final version.
ACKNOWLEDGEMENTS
The authors are grateful to the patient and his family for generously permitting usage of clinical information.
CONFLICT OF INTEREST
None of the authors have any conflict of interest to disclose.
FUNDING
No funding was obtained for this case report.
AVAILABILITY OF DATA
The data of the current case report are available and included in the manuscript.
CONSENT
The case report protocol was performed in accordance with the Declaration of Helsinki.
Loberti L, et al. Natural history of KBG syndrome in a large European cohort. Hum Mol Genet. 2022 Dec 16;31(24):4131-4142. View
at PublisherView
at Google Scholar
Auconi M, Serino D, Digilio MC, Gnazzo M, Conti M, Vigevano F, Fusco L. Epilepsy in KBG syndrome. Dev Med Child Neurol. 2023 May;65(5):712-720 View
at PublisherView
at Google Scholar
Buijsse N, et al. Epilepsy is an important feature of KBG syndrome associated with poorer developmental outcome. Epilepsia Open. 2023 Jul 27. View
at PublisherView
at Google Scholar
Miao P, Feng J, Guo Y, Wang J, Xu X, Wang Y, Li Y, Gao L, Zheng C, Cheng H. Genotype and phenotype analysis using an epilepsy-associated gene panel in Chinese pediatric epilepsy patients. Clin Genet. 2018 Dec;94(6):512-520. View
at PublisherView
at Google Scholar
Goldenberg A, et al. Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am J Med Genet A. 2016 Nov;170(11):2847-2859. View
at PublisherView
at Google Scholar
Low K, et al; DDD Study; Smithson S. Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. 2016 Nov;170(11):2835-2846 View
at PublisherView
at Google Scholar
Alves RM, et al. Novel ANKRD11 gene mutation in an individual with a mild phenotype of KBG syndrome associated to a GEFS+ phenotypic spectrum: a case report. BMC Med Genet. 2019 Jan 14;20(1):16. View
at PublisherView
at Google Scholar
Nardello R, Mangano GD, Antona V, Fontana A, Striano P, Giorgio E, Brusco A, Mangano S, Salpietro V. Electroclinical features and outcome of ANKRD11-related KBG syndrome: A novel report and literature review. Seizure. 2021 Feb; 85: 151-154. View
at PublisherView
at Google Scholar
Kutkowska-Kaźmierczak A, et al. Wide Fontanels, Delayed Speech Development and Hoarse Voice as Useful Signs in the Diagnosis of KBG Syndrome: A Clinical Description of 23 Cases with Pathogenic Variants Involving the ANKRD11Gene or Submicroscopic Chromosomal Rearrangements of 16q24.3. Genes (Basel). 2021 Aug 17;12(8):1257. View
at PublisherView
at Google Scholar
Kim SJ, Yang A, Park JS, Kwon DG, Lee JS, Kwon YS, Lee JE. Two Novel Mutations of ANKRD11 Gene and Wide Clinical Spectrum in KBG Syndrome: Case Reports and Literature Review. Front Genet. 2020 Nov 11; 11: 579805. View
at PublisherView
at Google Scholar
Murphy MJ, McSweeney N, Cavalleri GL, Greally MT, Benson KA, Costello DJ. KBG syndrome mimicking genetic generalized epilepsy. Epilepsy Behav Rep. 2022 Apr 20; 19: 100545. View
at PublisherView
at Google Scholar
Shneker BF, Fountain NB. Epilepsy. Dis Mon. 2003 Jul;49(7):426-78. Milligan TA. Epilepsy: A Clinical Overview. Am J Med. 2021 Jul;134(7):840-847. View
at PublisherView
at Google Scholar
Tang, F., Hartz, A., & Bauer, B. (2017). Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Frontiers in neurology, 8, 301. View
at PublisherView
at Google Scholar
Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, Resnick T, Benbadis SR. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014 Aug; 37:59-70. View
at PublisherView
at Google Scholar
Knupp, K., Koh, S., & Park, K. (2012). Pediatric epilepsy: Five new things. Neurology. Clinical practice, 2(1), 40–47. View
at PublisherView
at Google Scholar
"ScienceHood Publishing surpassed our expectations with their professionalism, timely communication, and exceptional attention to detail. They transformed our vision into reality with outstanding results. We highly recommend them for their expertise and commitment to excellence."
Shippora Smith
"ScienceHood Publishing exceeded our expectations with their seamless execution and professionalism. Their team ensured timely communication, high-quality production, and attention to detail throughout the process. They transformed our vision into reality, delivering exceptional results. We highly recommend them for their efficiency, expertise, and commitment to excellence in publishing."
Lara Simmons
""I had an exceptional experience publishing my research article with ScienceHood LLC. From the very first interaction, the team displayed utmost professionalism and provided prompt support throughout the entire process. The editorial team was thorough, offering constructive feedback that enhanced the quality of my work. Their attention to detail, timely communication, and dedication to scientific excellence truly set them apart. I am thrilled with the final publication, and I highly recommend ScienceHood LLC to fellow researchers seeking a reliable and proficient publishing platform.""
Paul Atkinson
"ScienceHood Publishing surpassed our expectations with their professionalism, timely communication, and exceptional attention to detail. They transformed our vision into reality with outstanding results. We highly recommend them for their expertise and commitment to excellence."
Shippora Smith
"ScienceHood Publishing exceeded our expectations with their seamless execution and professionalism. Their team ensured timely communication, high-quality production, and attention to detail throughout the process. They transformed our vision into reality, delivering exceptional results. We highly recommend them for their efficiency, expertise, and commitment to excellence in publishing."

 :
© 2025 Alili Ademi L. This open-access article is distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
:
© 2025 Alili Ademi L. This open-access article is distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.